Infantile fiber-type I hypotrophy with simultaneously occurring severe onset of cardiomyopathy was reported in multiple Dutch and Italian families and genetically linked to the MYL2 gene encoding for the human myosin regulatory light chain RLCventr/slow concurrently expressed in cardiac ventricles and in slow-twitch skeletal muscles. To address the molecular origin of this novel RLCventr/slow-induced cardioskeletal myopathy, we extensively investigate the functional consequences of hypertrophic cardiomyopathy (HCM)-linked mutations in MYL2 using cardiac papillary muscle fibers and slow-twitch skeletal soleus muscles from our developed transgenic (Tg) mice expressing RLC wild-type (Tg-WT) and FHC-linked mutants. Importantly, will also characterize the splice site mutations (e.g. IVS6-1) in MYL2 shown to cause severe myopathy in humans and premature death of IVS6-1-homozygous patients. The long term goal is to provide insight into molecular bases of FHC and a dual cardioskeletal myopathy caused by mutated RLCventr/slow.
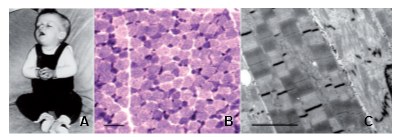
Clinical history and biopsy findings. (A) Patient N-A1 at 5 months: general limpness with facial palsy and ptosis. (B) Light microscopy of quadriceps muscle, NADH oxidoreductase reaction showing fiber-type disproportion. Smallest (dark) type I fibers show increased activity. Scale bar = 50 µm. (C) Electron microscopy of quadriceps muscle of Patient N-D1. Central fiber shows severe disarray of normal sarcomeric structure including Z-disks and lysis of myofibrils. Adjacent fibers show loss of register of sarcomeres. Scale bar = 1 µm. From Weterman et al. Brain 2013.